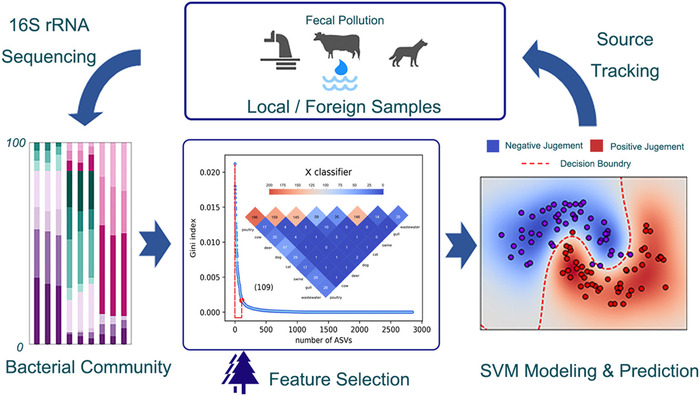
高通量测序所揭示的细菌多样性和相应的生物学意义为粪便污染源追踪提供了大量信息。文章应用支持向量机(support vector machine, SVM)算法,考察了分类模型在预测本地和外来样本粪便污染源方面的表现,并与随机森林(RF)和Adaboost进行比较。利用极随机树(ExtraTrees)从梭菌目(Clostridiale)、拟杆菌目(Bacteroidales)和乳杆菌目(Lactobacillales)细菌群中选取鉴别序列。在原始细菌文库中,约1.51-12.64%的独特序列构成了代表性生物学标记,贡献了源微生物组之间70%的差异。SVM模型和RF模型对本地样品的总体准确率分别为96.08%和98.04%,高于Adaboost(90.20%)。对于非本地样本,SVM成功将大部分粪便样本分配至正确的类别中,但在密切相关的类别中出现了一定的假阳性判断。本文的研究结果表明,SVM是一种省时、准确的污染水体粪源追踪方法,具有基于地理上不相关的样品执行任务的潜在能力,并且强调了qPCR分析对准确检测人源污染的必要性。
Abstract: The bacterial diversity andcorresponding biological significance revealed by high-throughput sequencingcontribute massive information to source tracking of fecal contamination. Theperformances of classification models on predicting the fecal source of geographicallocal and foreign samples were examined herein, by applying support vectormachine (SVM) algorithm. Random forest (RF) and Adaboost were applied forcomparison as well. Discriminatory sequences were selected from Clostridiale,Bacteroidales, or Lactobacillales bacterial groups using extremely randomizedtrees (ExtraTrees). 1.51–12.64% of the unique sequences in the original librarycomposed the representative markers, and they contributed 70% of thediscrepancies between source microbiomes. The overall accuracy of the SVM modeland the RF model on local samples was 96.08% and 98.04%, respectively, higherthan that of the Adaboost (90.20%). As for the non-local samples, the SVMassigned most of the fecal samples into the correct category while severalfalse-positive judgments occurred in closely related groups. The results inthis paper suggested that the SVM was a time-saving and accurate method forfecal source tracking in contaminated water body with the potential capabilityof executing tasks based on geographically unassociated samples, and underlinedthe necessity of qPCR analysis for accurate detection of human sourcepollution.
点击下载


